Cystic Fibrosis
What is Cystic Fibrosis?
Cystic fibrosis (CF) is a genetic condition increasingly recognized in the Indian subcontinent. It primarily affects the lungs and digestive system, leading to chronic health issues due to thick, sticky secretions.
What is the Genetic Defect in Cystic Fibrosis?
- The CFTR protein plays a crucial role in the production of sweat, digestive fluids, and mucus. When defective, it causes secretions to become thick and sticky, leading to blockages and damage to multiple organs.
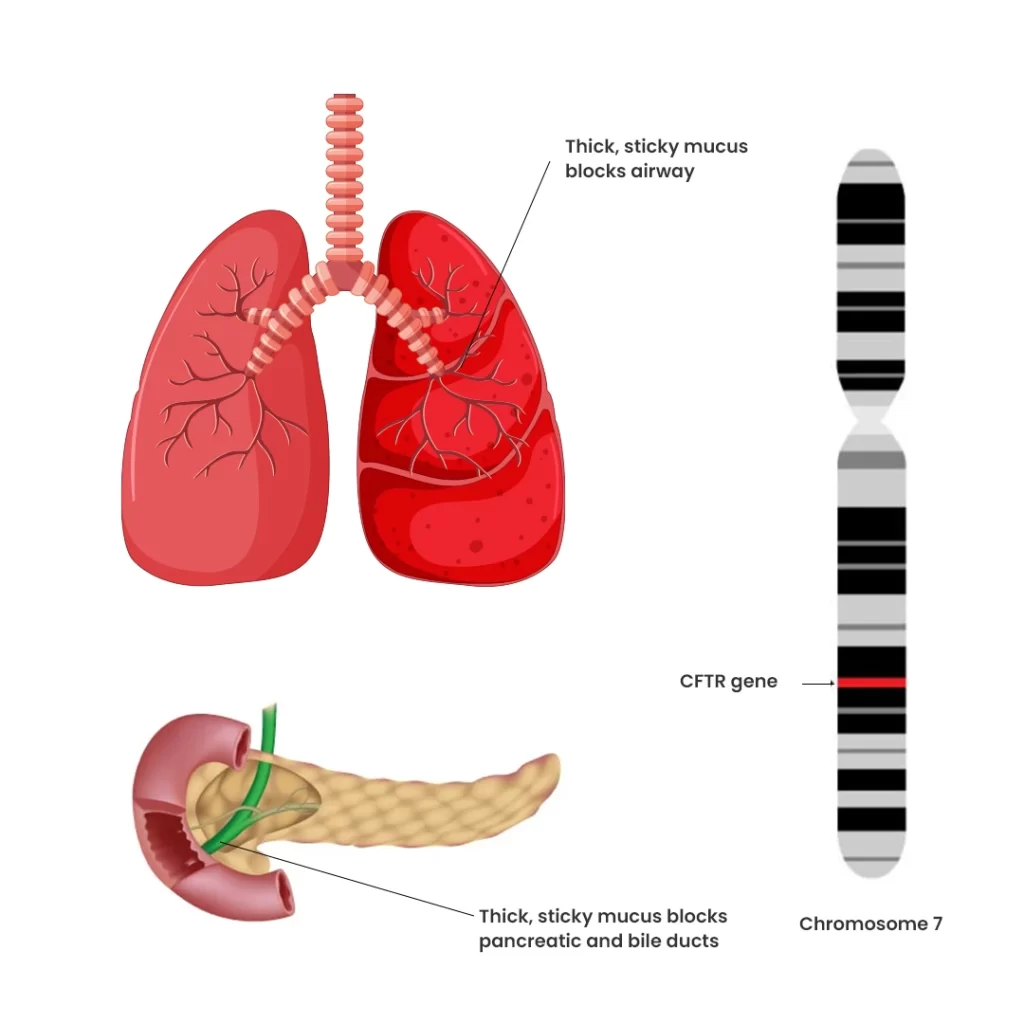
- CF is caused by mutations in the CFTR gene (Cystic Fibrosis Transmembrane Conductance Regulator). This condition is inherited in an autosomal recessive manner, meaning a child needs two copies of the defective gene—one from each parent—to develop the disease. Parents with a single copy of the defective gene are usually carriers and remain unaffected.
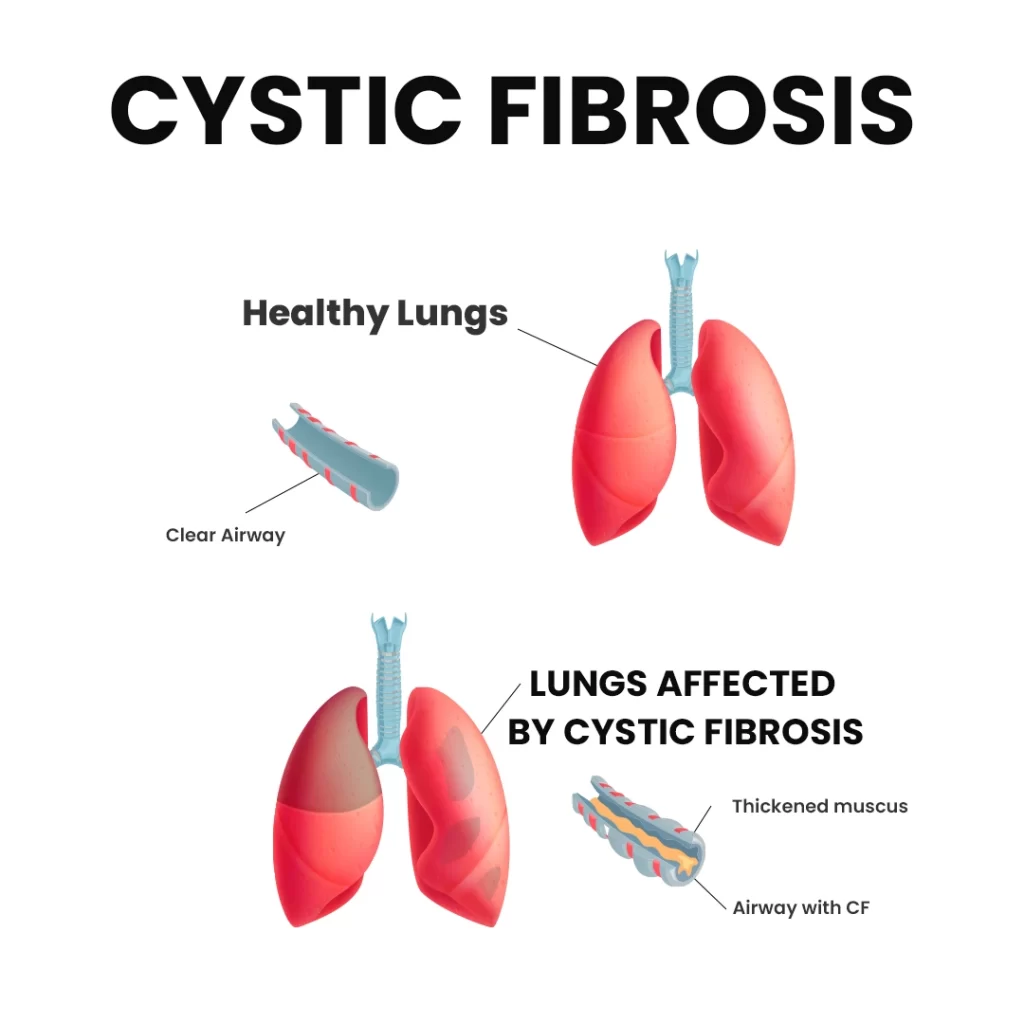
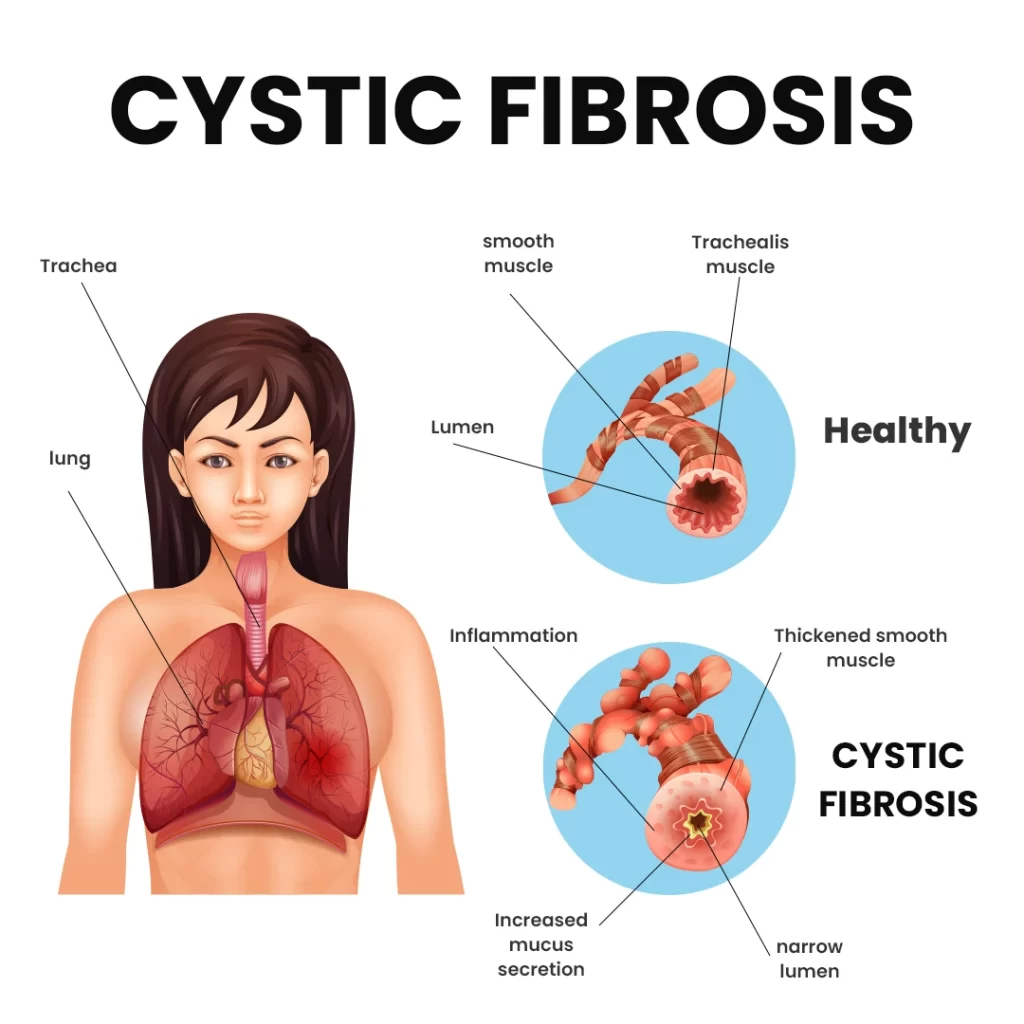
How Does Cystic Fibrosis Affect the Lungs?
The lungs are the primary organs impacted by CF. In affected children, mucus becomes thick and sticky, clogging the small airways and leading to recurrent infections and chronic lung damage from a young age.
What Other Organs Are Affected in Cystic Fibrosis?
- CF also affects the digestive system, including the liver, pancreas, and intestines. This leads to reduced production of digestive juices, impairing food absorption. As a result, children often have large, bulky, frothy stools, poor weight gain, and growth challenges.
Symptoms Suggestive of Cystic Fibrosis:
- Chronic cough with sputum production
- Wheezing and frequent lung infections (pneumonia or bronchitis)
- Persistent loose, oily stools
- Failure to thrive (difficulty gaining weight despite a good appetite)
- Poor growth
- Dehydration due to excessive salt loss in sweat

How is Cystic Fibrosis Diagnosed?
Diagnosis begins with a thorough review of symptoms and physical examination. If CF is suspected, key tests are performed:
- Sweat Chloride Analysis: The global standard test for CF diagnosis. A sweat sample is collected using Gibson & Cooke’s method, and a chloride level above 60 mmol/L is indicative of CF.
- Genetic Analysis: Clinical exome sequencing can confirm CFTR gene mutations.
- Sputum Test: Used to identify any lung microorganisms; if a sputum test is not possible, a bronchoscopy may be required.
Management and Follow-Up for CF Patients:
Long-term management includes regular follow-up with a pediatric pulmonologist. Treatment may involve:
- Regular chest physiotherapy
- Antibiotics to manage lung infections
- Inhaled hypertonic saline and anti-DNAase to thin mucus and improve airway clearance